Overlap-related operations
Overlap-related operations: cheatsheet
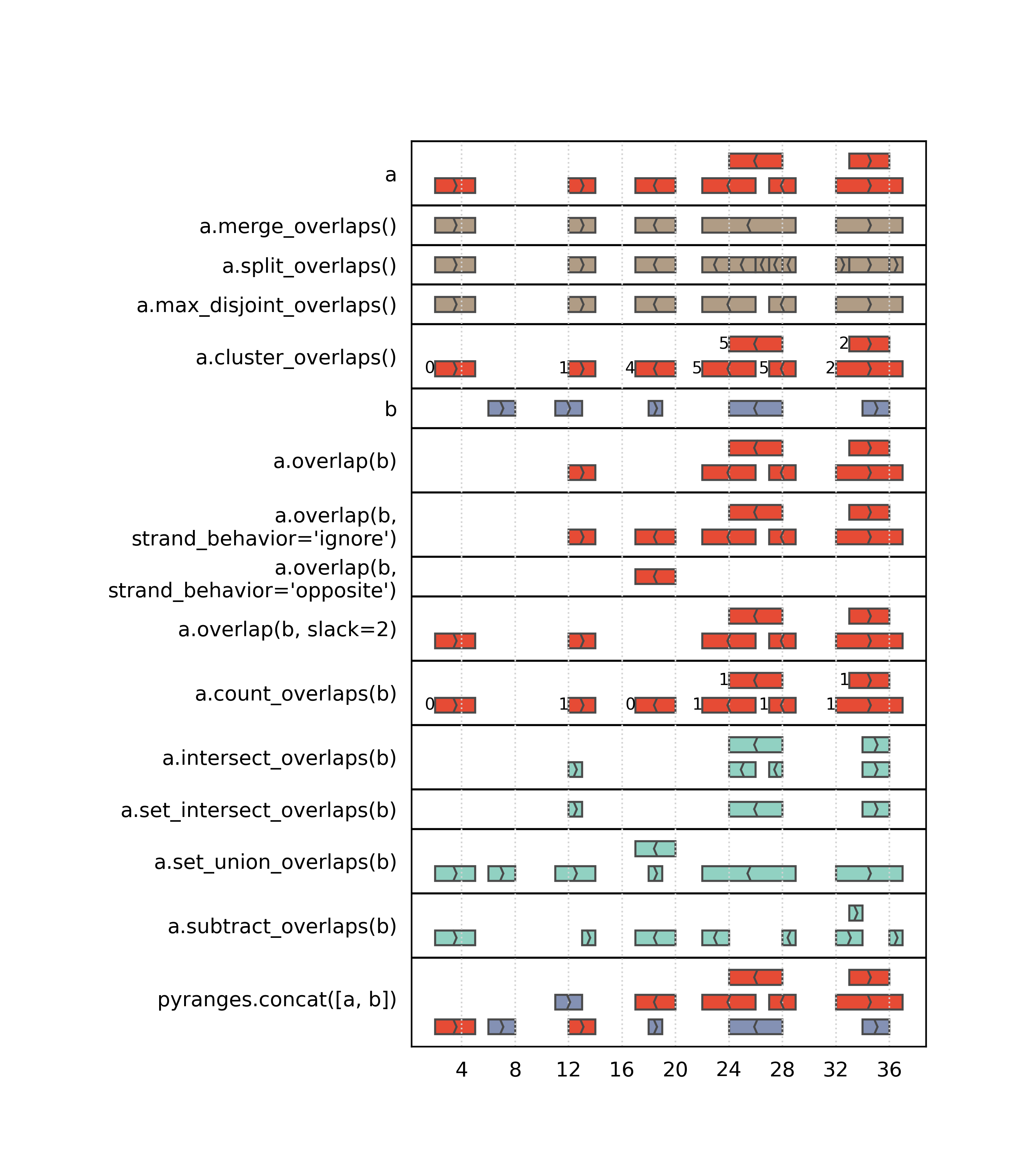
Single and pairwise PyRanges operations
Pyranges offers many efficient methods to detect / process overlaps. We present them here split in two groups:
Methods accepting a pair of PyRanges as input:
|
Return overlapping intervals. |
|
Join PyRanges based on genomic overlap. |
|
Return set-theoretical intersection. |
|
Return set-theoretical union. |
|
Return overlapping subintervals. |
|
Subtract intervals, i.e. return non-overlapping subintervals. |
|
Count number of overlaps per interval. |
|
Find closest interval. |
Methods accepting a single PyRanges as input:
|
Give overlapping intervals a common id. |
|
Merge overlapping intervals into one. |
|
Split into non-overlapping intervals. |
|
Find the maximal disjoint set of intervals. |
Methods for pairs of PyRanges
These methods accept two PyRanges objects as input (i.e. self and other), and return a new PyRanges object with the results of the operation.
We will briefly showcase them in this document. First, let’s create two PyRanges objects to work with:
>>> import pyranges1 as pr
>>> a = pr.PyRanges(dict(Chromosome="chr1",
... Start=[3, 13, 18, 23, 28, 32, 33],
... End=[6, 15, 21, 27, 29, 37, 36],
... Strand=["+", "+", "-", "-", "-", "+", "+"]))
>>> a
index | Chromosome Start End Strand
int64 | str int64 int64 str
------- --- ------------ ------- ------- --------
0 | chr1 3 6 +
1 | chr1 13 15 +
2 | chr1 18 21 -
3 | chr1 23 27 -
4 | chr1 28 29 -
5 | chr1 32 37 +
6 | chr1 33 36 +
PyRanges with 7 rows, 4 columns, and 1 index columns.
Contains 1 chromosomes and 2 strands.
>>> b = pr.PyRanges(dict(Chromosome="chr1",
... Start=[6, 12, 19, 25, 34],
... End=[8, 14, 20, 29, 36],
... Strand=["+", "+", "+", "-", "+"]))
>>> b
index | Chromosome Start End Strand
int64 | str int64 int64 str
------- --- ------------ ------- ------- --------
0 | chr1 6 8 +
1 | chr1 12 14 +
2 | chr1 19 20 +
3 | chr1 25 29 -
4 | chr1 34 36 +
PyRanges with 5 rows, 4 columns, and 1 index columns.
Contains 1 chromosomes and 2 strands.
Filter rows: overlap
The most intuitive overlap-related method is overlap.
This is simply a filter of the intervals in self, so that only those overlapping any interval in other are returned:
>>> a.overlap(b)
index | Chromosome Start End Strand
int64 | str int64 int64 str
------- --- ------------ ------- ------- --------
1 | chr1 13 15 +
3 | chr1 23 27 -
4 | chr1 28 29 -
5 | chr1 32 37 +
6 | chr1 33 36 +
PyRanges with 5 rows, 4 columns, and 1 index columns.
Contains 1 chromosomes and 2 strands.
Let’s now filter the rows of the other PyRanges object:
>>> b.overlap(a)
index | Chromosome Start End Strand
int64 | str int64 int64 str
------- --- ------------ ------- ------- --------
1 | chr1 12 14 +
3 | chr1 25 29 -
4 | chr1 34 36 +
PyRanges with 3 rows, 4 columns, and 1 index columns.
Contains 1 chromosomes and 2 strands.
Note above how intervals overlapping with more than one interval in the other PyRanges are not reported multiple times.
Use multiple=True to do so.
To get the self intervals without overlap in other, use invert=True:
>>> a.overlap(b, invert=True)
index | Chromosome Start End Strand
int64 | str int64 int64 str
------- --- ------------ ------- ------- --------
0 | chr1 3 6 +
2 | chr1 18 21 -
PyRanges with 2 rows, 4 columns, and 1 index columns.
Contains 1 chromosomes and 2 strands.
A reminder that intervals are encoded in pythonic convention: 0-based coordinates, with start included and end excluded. Thus, the closest possible intervals that are not overlapping are two intervals wherein the end of the first is equal of the start of the second. These are called “bookended” intervals, e.g.:
>>> a.head(1)
index | Chromosome Start End Strand
int64 | str int64 int64 str
------- --- ------------ ------- ------- --------
0 | chr1 3 6 +
PyRanges with 1 rows, 4 columns, and 1 index columns.
Contains 1 chromosomes and 1 strands.
>>> b.head(1)
index | Chromosome Start End Strand
int64 | str int64 int64 str
------- --- ------------ ------- ------- --------
0 | chr1 6 8 +
PyRanges with 1 rows, 4 columns, and 1 index columns.
Contains 1 chromosomes and 1 strands.
Common arguments: slack, strand_behavior, match_by
We will now use overlap to showcase arguments
that are available in many overlap-related methods.
slack (default: 0) is used to relax the criteria of overlap.
A value of 1 will report bookended intervals, previously not considered overlapping:
>>> b.overlap(a, slack=1)
index | Chromosome Start End Strand
int64 | str int64 int64 str
------- --- ------------ ------- ------- --------
0 | chr1 6 8 +
1 | chr1 12 14 +
3 | chr1 25 29 -
4 | chr1 34 36 +
PyRanges with 4 rows, 4 columns, and 1 index columns.
Contains 1 chromosomes and 2 strands.
Analogously, higher values will report increasingly distant intervals. In practice, the self intervals are temporarily extended by the slack amount on both ends before the overlap operation.
strand_behavior determines how strand is treated.
The value ‘same’ results in the intuitive behavior, i.e. two intervals overlap only if on the same strand:
>>> b.overlap(a, strand_behavior="same") # the result here is the same as: a.overlap(b)
index | Chromosome Start End Strand
int64 | str int64 int64 str
------- --- ------------ ------- ------- --------
1 | chr1 12 14 +
3 | chr1 25 29 -
4 | chr1 34 36 +
PyRanges with 3 rows, 4 columns, and 1 index columns.
Contains 1 chromosomes and 2 strands.
On the other hand, value ‘ignore’ will define overlaps regardless of strands:
>>> a.overlap(b, strand_behavior="ignore")
index | Chromosome Start End Strand
int64 | str int64 int64 str
------- --- ------------ ------- ------- --------
1 | chr1 13 15 +
2 | chr1 18 21 -
3 | chr1 23 27 -
4 | chr1 28 29 -
5 | chr1 32 37 +
6 | chr1 33 36 +
PyRanges with 6 rows, 4 columns, and 1 index columns.
Contains 1 chromosomes and 2 strands.
Value ‘opposite’ will require two intervals to be on the opposite strands to be considered overlapping:
>>> a.overlap(b, strand_behavior="opposite")
index | Chromosome Start End Strand
int64 | str int64 int64 str
------- --- ------------ ------- ------- --------
2 | chr1 18 21 -
PyRanges with 1 rows, 4 columns, and 1 index columns.
Contains 1 chromosomes and 1 strands.
Naturally, values ‘same’ and ‘opposite’ can only be used when the PyRanges objects have valid strand information, i.e. the Strand column is present and all its values are either ‘+’ or ‘-‘:
>>> b_unstranded = b.remove_strand()
>>> b_unstranded
index | Chromosome Start End
int64 | str int64 int64
------- --- ------------ ------- -------
0 | chr1 6 8
1 | chr1 12 14
2 | chr1 19 20
3 | chr1 25 29
4 | chr1 34 36
PyRanges with 5 rows, 3 columns, and 1 index columns.
Contains 1 chromosomes.
>>> a.overlap(b_unstranded, strand_behavior="same")
Traceback (most recent call last):
...
ValueError: Can only do same strand operations when both PyRanges contain valid strand info.
See function strand_valid for details, and
make_strand_valid to convert non-standard strand values to standard ones.
The default value of strand_behavior is ‘auto’.
This is transformed to ‘same’ if both PyRanges have valid strands, and to ‘ignore’ otherwise:
>>> a.overlap(b).equals(
... a.overlap(b, strand_behavior='same') )
True
>>> a.overlap(b_unstranded).equals(
... a.overlap(b, strand_behavior='ignore') )
True
Above, we leveraged method equals inherited from pandas Dataframe to compare table contents.
Note that the presence of any non-standard Strand value will result in strand being ignored for all rows. When leading to potentially non-intuitive behavior, a warning is printed:
>>> a_invalid = a.copy()
>>> a_invalid.loc[2, 'Strand'] = "."
>>> a_invalid
index | Chromosome Start End Strand
int64 | str int64 int64 str
------- --- ------------ ------- ------- --------
0 | chr1 3 6 +
1 | chr1 13 15 +
2 | chr1 18 21 .
3 | chr1 23 27 -
4 | chr1 28 29 -
5 | chr1 32 37 +
6 | chr1 33 36 +
PyRanges with 7 rows, 4 columns, and 1 index columns.
Contains 1 chromosomes and 3 strands (including non-genomic strands: .).
>>> a_invalid.overlap(b)
<input>:1: UserWarning: overlap: 'auto' strand_behavior treated as ignore due to invalid Strand values. Please use strand_behavior=ignore
index | Chromosome Start End Strand
int64 | str int64 int64 str
------- --- ------------ ------- ------- --------
1 | chr1 13 15 +
2 | chr1 18 21 .
3 | chr1 23 27 -
4 | chr1 28 29 -
5 | chr1 32 37 +
6 | chr1 33 36 +
PyRanges with 6 rows, 4 columns, and 1 index columns.
Contains 1 chromosomes and 3 strands (including non-genomic strands: .).
Finally, argument match_by can be used to specify additional columns whose values must match for two intervals
to be considered overlapping.
For example, let’s add a column to both objects to mark intervals whose Start is an odd number, then
use this column to filter the overlaps:
>>> a2 = a.assign(odd = lambda x:x.Start % 2 )
>>> a2
index | Chromosome Start End Strand odd
int64 | str int64 int64 str int64
------- --- ------------ ------- ------- -------- -------
0 | chr1 3 6 + 1
1 | chr1 13 15 + 1
2 | chr1 18 21 - 0
3 | chr1 23 27 - 1
4 | chr1 28 29 - 0
5 | chr1 32 37 + 0
6 | chr1 33 36 + 1
PyRanges with 7 rows, 5 columns, and 1 index columns.
Contains 1 chromosomes and 2 strands.
>>> b2 = b.assign(odd = lambda x:x.Start % 2 )
>>> b2
index | Chromosome Start End Strand odd
int64 | str int64 int64 str int64
------- --- ------------ ------- ------- -------- -------
0 | chr1 6 8 + 0
1 | chr1 12 14 + 0
2 | chr1 19 20 + 1
3 | chr1 25 29 - 1
4 | chr1 34 36 + 0
PyRanges with 5 rows, 5 columns, and 1 index columns.
Contains 1 chromosomes and 2 strands.
>>> a2.overlap(b2, match_by='odd')
index | Chromosome Start End Strand odd
int64 | str int64 int64 str int64
------- --- ------------ ------- ------- -------- -------
3 | chr1 23 27 - 1
5 | chr1 32 37 + 0
PyRanges with 2 rows, 5 columns, and 1 index columns.
Contains 1 chromosomes and 2 strands.
The most versatile: join_overlaps
The most versatile overlap-related method for pairs of PyRanges is join_overlaps.
This method is analogous to a SQL join operation, but rather than matching rows in two tables through a common key,
they are matched in virtue of their overlap.
This function searches for overlaps between the intervals in self and other, and reports in output the full information related to the input intervals. The returned PyRanges object will have a number of rows equal to the number of overlaps found, and the columns will be the union of the columns of self and other, using a suffix to differentiate columns in other which are present with the same name in the self PyRanges, like Start and End:
>>> a.join_overlaps(b)
index | Chromosome Start End Strand Start_b End_b
int64 | str int64 int64 str int64 int64
------- --- ------------ ------- ------- -------- --------- -------
1 | chr1 13 15 + 12 14
3 | chr1 23 27 - 25 29
4 | chr1 28 29 - 25 29
5 | chr1 32 37 + 34 36
6 | chr1 33 36 + 34 36
PyRanges with 5 rows, 6 columns, and 1 index columns.
Contains 1 chromosomes and 2 strands.
In contrast to overlap, a row is returned per overlap, so
if an interval in self overlaps with more than one interval in other, it will be reported multiple times,
resulting in index duplicates (see below); you may call .reset_index() to correct this.
>>> b.join_overlaps(a)
index | Chromosome Start End Strand Start_b End_b
int64 | str int64 int64 str int64 int64
------- --- ------------ ------- ------- -------- --------- -------
1 | chr1 12 14 + 13 15
3 | chr1 25 29 - 23 27
3 | chr1 25 29 - 28 29
4 | chr1 34 36 + 32 37
4 | chr1 34 36 + 33 36
PyRanges with 5 rows, 6 columns, and 1 index columns (with 2 index duplicates).
Contains 1 chromosomes and 2 strands.
Like all overlap-related methods accepting two PyRanges as input,
join_overlaps
accepts the strand_behavior argument. If Strand is not used to determine overlaps, it will
be returned for both PyRanges:
>>> a.join_overlaps(b, strand_behavior="ignore")
index | Chromosome Start End Strand Start_b End_b Strand_b
int64 | str int64 int64 str int64 int64 str
------- --- ------------ ------- ------- -------- --------- ------- ----------
1 | chr1 13 15 + 12 14 +
2 | chr1 18 21 - 19 20 +
3 | chr1 23 27 - 25 29 -
4 | chr1 28 29 - 25 29 -
5 | chr1 32 37 + 34 36 +
6 | chr1 33 36 + 34 36 +
PyRanges with 6 rows, 7 columns, and 1 index columns.
Contains 1 chromosomes and 2 strands.
slack and match_by arguments are also available, e.g.:
>>> a2.join_overlaps(b2, match_by='odd')
index | Chromosome Start End Strand odd Start_b End_b
int64 | str int64 int64 str int64 int64 int64
------- --- ------------ ------- ------- -------- ------- --------- -------
3 | chr1 23 27 - 1 25 29
5 | chr1 32 37 + 0 34 36
PyRanges with 2 rows, 7 columns, and 1 index columns.
Contains 1 chromosomes and 2 strands.
>>> a2.join_overlaps(b2, match_by='odd', slack=5)
index | Chromosome Start End Strand odd Start_b End_b
int64 | str int64 int64 str int64 int64 int64
------- --- ------------ ------- ------- -------- ------- --------- -------
1 | chr1 13 15 + 1 19 20
3 | chr1 23 27 - 1 25 29
5 | chr1 32 37 + 0 34 36
PyRanges with 3 rows, 7 columns, and 1 index columns.
Contains 1 chromosomes and 2 strands.
Pyranges provides method combine_interval_columns
to post-process the output of join_overlaps
and aggregate the coordinates of the overlapping intervals in Start and End columns.
For example, this allows to obtain the union of the overlapping intervals:
>>> a2.join_overlaps(b2, match_by='odd', slack=5).combine_interval_columns('union')
index | Chromosome Start End Strand odd
int64 | str int64 int64 str int64
------- --- ------------ ------- ------- -------- -------
1 | chr1 13 20 + 1
3 | chr1 23 29 - 1
5 | chr1 32 37 + 0
PyRanges with 3 rows, 5 columns, and 1 index columns.
Contains 1 chromosomes and 2 strands.
join_overlaps is one of most versatile methods in PyRanges, since it
retains the full information of the two input PyRanges objects.
Nevertheless, more efficient alternative methods are available for specific use cases, detailed below.
Set operations: set_intersect_overlaps, set_union_overlaps
Pyranges offers efficient methods based on the concept of set operations in mathematics. These are useful when the user is interested in the intervals themselves, rather than in the full information (i.e. metadata) of the input intervals.
Method set_intersect_overlaps, allows to obtain the genomic regions
present in both PyRanges:
>>> a.set_intersect_overlaps(b)
index | Chromosome Start End Strand
int64 | str int64 int64 str
------- --- ------------ ------- ------- --------
0 | chr1 13 14 +
1 | chr1 34 36 +
2 | chr1 25 27 -
3 | chr1 28 29 -
PyRanges with 4 rows, 4 columns, and 1 index columns.
Contains 1 chromosomes and 2 strands.
>>> a.set_intersect_overlaps(b, strand_behavior="ignore")
index | Chromosome Start End
int64 | str int64 int64
------- --- ------------ ------- -------
0 | chr1 13 14
1 | chr1 19 20
2 | chr1 25 27
3 | chr1 28 29
4 | chr1 34 36
PyRanges with 5 rows, 3 columns, and 1 index columns.
Contains 1 chromosomes.
The regions reported may be part of any interval in the two PyRanges. All metadata columns are dropped:
>>> a2.set_intersect_overlaps(b2).columns # see above: a2 and b2 had the 'odd' column
Index(['Chromosome', 'Start', 'End', 'Strand'], dtype='str')
Analogously, method set_union_overlaps allows to obtain the genomic regions that
are present in at least one of the PyRanges:
>>> a.set_union_overlaps(b)
index | Chromosome Start End Strand
int64 | str int64 int64 str
------- --- ------------ ------- ------- --------
0 | chr1 3 6 +
1 | chr1 6 8 +
2 | chr1 12 15 +
3 | chr1 19 20 +
4 | chr1 32 37 +
5 | chr1 18 21 -
6 | chr1 23 29 -
PyRanges with 7 rows, 4 columns, and 1 index columns.
Contains 1 chromosomes and 2 strands.
>>> a2.set_union_overlaps(b2, strand_behavior='ignore')
index | Chromosome Start End
int64 | str int64 int64
------- --- ------------ ------- -------
0 | chr1 3 6
1 | chr1 6 8
2 | chr1 12 15
3 | chr1 18 21
4 | chr1 23 29
5 | chr1 32 37
PyRanges with 6 rows, 3 columns, and 1 index columns.
Contains 1 chromosomes.
Interval manipulation operations: intersect_overlaps, subtract_overlaps
Set operations do not preserve input metadata.
join_overlaps preserve metadata of both PyRanges, but is less efficient.
Pyranges also offers methods that preserve the metadata in self, but not in other.
Specifically, method intersect_overlaps allows to obtain the intervals in self that overlap
with any interval in other. It is similar to overlap, but here coordinates
are modified to return only the actual overlaps:
>>> a2.intersect_overlaps(b)
index | Chromosome Start End Strand odd
int64 | str int64 int64 str int64
------- --- ------------ ------- ------- -------- -------
1 | chr1 13 14 + 1
3 | chr1 25 27 - 1
4 | chr1 28 29 - 0
5 | chr1 34 36 + 0
6 | chr1 34 36 + 1
PyRanges with 5 rows, 5 columns, and 1 index columns.
Contains 1 chromosomes and 2 strands.
>>> a2.intersect_overlaps(b2, strand_behavior='ignore', match_by='odd')
index | Chromosome Start End Strand odd
int64 | str int64 int64 str int64
------- --- ------------ ------- ------- -------- -------
3 | chr1 25 27 - 1
5 | chr1 34 36 + 0
PyRanges with 2 rows, 5 columns, and 1 index columns.
Contains 1 chromosomes and 2 strands.
Method subtract_overlaps allows to obtain the portions of intervals in self
that do not overlap any interval in other:
>>> a2.subtract_overlaps(b).sort_values('Start')
index | Chromosome Start End Strand odd
int64 | str int64 int64 str int64
------- --- ------------ ------- ------- -------- -------
0 | chr1 3 6 + 1
1 | chr1 14 15 + 1
2 | chr1 18 21 - 0
3 | chr1 23 25 - 1
5 | chr1 32 34 + 0
6 | chr1 33 34 + 1
5 | chr1 36 37 + 0
PyRanges with 7 rows, 5 columns, and 1 index columns (with 1 index duplicates).
Contains 1 chromosomes and 2 strands.
>>> a2.subtract_overlaps(b, strand_behavior='ignore').sort_values('Start')
index | Chromosome Start End Strand odd
int64 | str int64 int64 str int64
------- --- ------------ ------- ------- -------- -------
0 | chr1 3 6 + 1
1 | chr1 14 15 + 1
2 | chr1 18 19 - 0
2 | chr1 20 21 - 0
3 | chr1 23 25 - 1
5 | chr1 32 34 + 0
6 | chr1 33 34 + 1
5 | chr1 36 37 + 0
PyRanges with 8 rows, 5 columns, and 1 index columns (with 2 index duplicates).
Contains 1 chromosomes and 2 strands.
Fast counting: count_overlaps
Method count_overlaps allows to count, for each interval in self,
the number of intervals in other that overlaps with it.
Input coordinates are not modified, and a new column is added:
>>> a2.count_overlaps(b) # using a2 to show the 'odd' column is preserved
index | Chromosome Start End Strand odd Count
int64 | str int64 int64 str int64 uint32
------- --- ------------ ------- ------- -------- ------- --------
0 | chr1 3 6 + 1 0
1 | chr1 13 15 + 1 1
2 | chr1 18 21 - 0 0
3 | chr1 23 27 - 1 1
4 | chr1 28 29 - 0 1
5 | chr1 32 37 + 0 1
6 | chr1 33 36 + 1 1
PyRanges with 7 rows, 6 columns, and 1 index columns.
Contains 1 chromosomes and 2 strands.
Arguments strand_behavior and match_by are available:
>>> a.count_overlaps(b, strand_behavior='ignore')
index | Chromosome Start End Strand Count
int64 | str int64 int64 str uint32
------- --- ------------ ------- ------- -------- --------
0 | chr1 3 6 + 0
1 | chr1 13 15 + 1
2 | chr1 18 21 - 1
3 | chr1 23 27 - 1
4 | chr1 28 29 - 1
5 | chr1 32 37 + 1
6 | chr1 33 36 + 1
PyRanges with 7 rows, 5 columns, and 1 index columns.
Contains 1 chromosomes and 2 strands.
>>> a2.count_overlaps(b2, strand_behavior='ignore', match_by='odd')
index | Chromosome Start End Strand odd Count
int64 | str int64 int64 str int64 uint32
------- --- ------------ ------- ------- -------- ------- --------
0 | chr1 3 6 + 1 0
1 | chr1 13 15 + 1 0
2 | chr1 18 21 - 0 0
3 | chr1 23 27 - 1 1
4 | chr1 28 29 - 0 0
5 | chr1 32 37 + 0 1
6 | chr1 33 36 + 1 0
PyRanges with 7 rows, 6 columns, and 1 index columns.
Contains 1 chromosomes and 2 strands.
Advanced metrics (e.g. interval fraction covered by overlap)
can be computed using the compute_interval_metrics
function, which is run on the output of join_overlaps:
>>> a.join_overlaps(b, strand_behavior='ignore').compute_interval_metrics('fraction')
index | Chromosome Start End Strand Start_b End_b Strand_b fraction
int64 | str int64 int64 str int64 int64 str float64
------- --- ------------ ------- ------- -------- --------- ------- ---------- ----------
1 | chr1 13 15 + 12 14 + 0.5
2 | chr1 18 21 - 19 20 + 0.333333
3 | chr1 23 27 - 25 29 - 0.5
4 | chr1 28 29 - 25 29 - 1
5 | chr1 32 37 + 34 36 + 0.4
6 | chr1 33 36 + 34 36 + 0.666667
PyRanges with 6 rows, 8 columns, and 1 index columns.
Contains 1 chromosomes and 2 strands.
Find the closest interval: nearest_ranges
Method nearest_ranges allows to find the closest interval in other for each interval
in self:
>>> a.nearest_ranges(b)
index | Chromosome Start End Strand Chromosome_b Start_b End_b Strand_b Distance
int64 | str int64 int64 str str int64 int64 str int64
------- --- ------------ ------- ------- -------- -------------- --------- ------- ---------- ----------
0 | chr1 3 6 + chr1 6 8 + 1
1 | chr1 13 15 + chr1 12 14 + 0
2 | chr1 18 21 - chr1 25 29 - 5
3 | chr1 23 27 - chr1 25 29 - 0
4 | chr1 28 29 - chr1 25 29 - 0
5 | chr1 32 37 + chr1 34 36 + 0
6 | chr1 33 36 + chr1 34 36 + 0
PyRanges with 7 rows, 9 columns, and 1 index columns.
Contains 1 chromosomes and 2 strands.
The output format is similar to join_overlaps.
Note the “Distance” column, which reports the distance between the intervals in self and other.
In case you want to find the nearest interval which does not overlap with each self interval, use
exclude_overlaps=True:
>>> a.nearest_ranges(b, exclude_overlaps=True)
index | Chromosome Start End Strand Chromosome_b Start_b End_b Strand_b Distance
int64 | str int64 int64 str str int64 int64 str int64
------- --- ------------ ------- ------- -------- -------------- --------- ------- ---------- ----------
0 | chr1 3 6 + chr1 6 8 + 1
1 | chr1 13 15 + chr1 19 20 + 5
2 | chr1 18 21 - chr1 25 29 - 5
5 | chr1 32 37 + chr1 19 20 + 13
6 | chr1 33 36 + chr1 19 20 + 14
PyRanges with 5 rows, 9 columns, and 1 index columns.
Contains 1 chromosomes and 2 strands.
The nearest_ranges method also accepts the strand_behavior argument:
>>> a.nearest_ranges(b, strand_behavior='ignore', exclude_overlaps=True)
index | Chromosome Start End Strand Chromosome_b Start_b End_b Strand_b Distance
int64 | str int64 int64 str str int64 int64 str int64
------- --- ------------ ------- ------- -------- -------------- --------- ------- ---------- ----------
0 | chr1 3 6 + chr1 6 8 + 1
1 | chr1 13 15 + chr1 19 20 + 5
2 | chr1 18 21 - chr1 12 14 + 5
2 | chr1 18 21 - chr1 25 29 - 5
3 | chr1 23 27 - chr1 19 20 + 4
4 | chr1 28 29 - chr1 34 36 + 6
5 | chr1 32 37 + chr1 25 29 - 4
6 | chr1 33 36 + chr1 25 29 - 5
PyRanges with 8 rows, 9 columns, and 1 index columns (with 1 index duplicates).
Contains 1 chromosomes and 2 strands.
Methods for single PyRanges
These overlap-related methods return a modified version of the input PyRanges object (self). We will showcase them with this data:
>>> c = pr.PyRanges(dict(Chromosome="chr1",
... Start=[1, 4, 10, 12, 19, 20, 24, 28],
... End=[5, 7, 14, 16, 27, 22, 25, 30],
... Strand=["+", "+", "+", "-", "+", "+", "+", "+"]))
>>> c
index | Chromosome Start End Strand
int64 | str int64 int64 str
------- --- ------------ ------- ------- --------
0 | chr1 1 5 +
1 | chr1 4 7 +
2 | chr1 10 14 +
3 | chr1 12 16 -
4 | chr1 19 27 +
5 | chr1 20 22 +
6 | chr1 24 25 +
7 | chr1 28 30 +
PyRanges with 8 rows, 4 columns, and 1 index columns.
Contains 1 chromosomes and 2 strands.
Grouping overlapping intervals: cluster_overlaps
The most flexible method in this category is cluster_overlaps.
This function will detect overlaps among intervals in self, and assign a cluster identifier
to each group of overlapping intervals. The object returned is identical to the input,
with an additional column “Cluster” containing the cluster identifier:
>>> c.cluster_overlaps()
index | Chromosome Start End Strand Cluster
int64 | str int64 int64 str uint32
------- --- ------------ ------- ------- -------- ---------
0 | chr1 1 5 + 0
1 | chr1 4 7 + 0
2 | chr1 10 14 + 1
4 | chr1 19 27 + 2
5 | chr1 20 22 + 2
6 | chr1 24 25 + 2
7 | chr1 28 30 + 3
3 | chr1 12 16 - 5
PyRanges with 8 rows, 5 columns, and 1 index columns.
Contains 1 chromosomes and 2 strands.
Note that clusters 1 and 2 are kept separated only because of strand.
We introduce argument use_strand, accepted by all overlap-related methods for single PyRanges.
When set to False, strand is ignored for overlap detection:
>>> c.cluster_overlaps(use_strand=False)
index | Chromosome Start End Strand Cluster
int64 | str int64 int64 str uint32
------- --- ------------ ------- ------- -------- ---------
0 | chr1 1 5 + 0
1 | chr1 4 7 + 0
2 | chr1 10 14 + 1
3 | chr1 12 16 - 1
4 | chr1 19 27 + 2
5 | chr1 20 22 + 2
6 | chr1 24 25 + 2
7 | chr1 28 30 + 3
PyRanges with 8 rows, 5 columns, and 1 index columns.
Contains 1 chromosomes and 2 strands.
The default value of argument use_strand is ‘auto’, which is interpreted as True if the PyRanges object
has valid strand information, and False otherwise.
Note that cluster 2 contain intervals with indices 5 and 6 which do not directly overlap, but they both overlap with the interval with index 4. (More generally, a cluster is a connected component of the overlap graph, where two intervals are connected if they overlap directly.)
Argument cluster_column controls the name of the column containing the cluster identifier.
Also, argument slack is also available here, and it is analogous to its use in methods for pairs of PyRanges.
Its default value is 0. With slack=1, bookended intervals are placed in the same cluster.
With slack=2, intervals that are distant at the most 1 bp are placed in the same cluster,
like those with index 4 and 7 above; and so on.
>>> c2 = c.cluster_overlaps(slack=2, use_strand=False, cluster_column='myClust')
>>> c2
index | Chromosome Start End Strand myClust
int64 | str int64 int64 str uint32
------- --- ------------ ------- ------- -------- ---------
0 | chr1 1 5 + 0
1 | chr1 4 7 + 0
2 | chr1 10 14 + 1
3 | chr1 12 16 - 1
4 | chr1 19 27 + 2
5 | chr1 20 22 + 2
6 | chr1 24 25 + 2
7 | chr1 28 30 + 2
PyRanges with 8 rows, 5 columns, and 1 index columns.
Contains 1 chromosomes and 2 strands.
Argument match_by is also available.
Only intervals with the same value in the specified column will be considered for overlap detection.
Let’s add a gene column to the PyRanges object and compare “Cluster” results with the previous column “myClust”:
>>> c2['gene'] = ['abc'[s % 3] for s in c2.Start] # arbitrary gene assignment
>>> c2.cluster_overlaps(slack=2, use_strand=False, match_by='gene')
index | Chromosome Start End Strand myClust gene Cluster
int64 | str int64 int64 str uint32 str uint32
------- --- ------------ ------- ------- -------- --------- ------ ---------
3 | chr1 12 16 - 1 a 0
6 | chr1 24 25 + 2 a 1
0 | chr1 1 5 + 0 b 3
1 | chr1 4 7 + 0 b 3
2 | chr1 10 14 + 1 b 4
4 | chr1 19 27 + 2 b 5
7 | chr1 28 30 + 2 b 5
5 | chr1 20 22 + 2 c 7
PyRanges with 8 rows, 7 columns, and 1 index columns.
Contains 1 chromosomes and 2 strands.
Resolve overlaps: merge_overlaps, split_overlaps, max_disjoint_overlaps
Various methods exists to obtain a PyRanges object without internal overlaps.
Method merge_overlaps
allows to merge overlapping intervals in self.
In practice, this function returns the union of all intervals in self.
All metadata columns are dropped:
>>> c.merge_overlaps()
index | Chromosome Start End Strand
int64 | str int64 int64 str
------- --- ------------ ------- ------- --------
0 | chr1 1 7 +
1 | chr1 10 14 +
2 | chr1 19 27 +
3 | chr1 28 30 +
4 | chr1 12 16 -
PyRanges with 5 rows, 4 columns, and 1 index columns.
Contains 1 chromosomes and 2 strands.
As before, both slack and use_strand are supported:
>>> c.merge_overlaps(slack=2)
index | Chromosome Start End Strand
int64 | str int64 int64 str
------- --- ------------ ------- ------- --------
0 | chr1 1 7 +
1 | chr1 10 14 +
2 | chr1 19 30 +
3 | chr1 12 16 -
PyRanges with 4 rows, 4 columns, and 1 index columns.
Contains 1 chromosomes and 2 strands.
Note that Strand is not reported when use_strand is set to False:
>>> c.merge_overlaps(slack=2, use_strand=False)
index | Chromosome Start End
int64 | str int64 int64
------- --- ------------ ------- -------
0 | chr1 1 7
1 | chr1 10 16
2 | chr1 19 30
PyRanges with 3 rows, 3 columns, and 1 index columns.
Contains 1 chromosomes.
On the other hand, method split_overlaps allows
to split intervals in self
at the position of overlaps, leaving as many bookended intervals as necessary to avoid overlaps.
This function drops metadata, too:
>>> pr.options.set_option('max_rows_to_show', 12) # to see all rows
>>> c.split_overlaps()
index | Chromosome Start End Strand
int64 | str int64 int64 str
------- --- ------------ ------- ------- --------
0 | chr1 1 4 +
1 | chr1 4 5 +
2 | chr1 5 7 +
3 | chr1 10 14 +
4 | chr1 19 20 +
5 | chr1 20 22 +
6 | chr1 22 24 +
7 | chr1 24 25 +
8 | chr1 25 27 +
9 | chr1 28 30 +
10 | chr1 12 16 -
PyRanges with 11 rows, 4 columns, and 1 index columns.
Contains 1 chromosomes and 2 strands.
Function max_disjoint_overlaps also returns a set of non-overlapping intervals.
In this case, however, input intervals are not modified, just filtered.
The intervals to return are chosen to maximize the number of intervals in the output.
The usual arguments (e.g. use_strand) are available:
>>> pr.options.reset_options()
>>> c2.max_disjoint_overlaps(use_strand=False) # using c2 to show that metadata is retained
index | Chromosome Start End Strand myClust gene
int64 | str int64 int64 str uint32 str
------- --- ------------ ------- ------- -------- --------- ------
0 | chr1 1 5 + 0 b
2 | chr1 10 14 + 1 b
4 | chr1 19 27 + 2 b
7 | chr1 28 30 + 2 b
PyRanges with 4 rows, 6 columns, and 1 index columns.
Contains 1 chromosomes and 1 strands.