Operating on coordinates
Operating on coordinates: cheatsheet
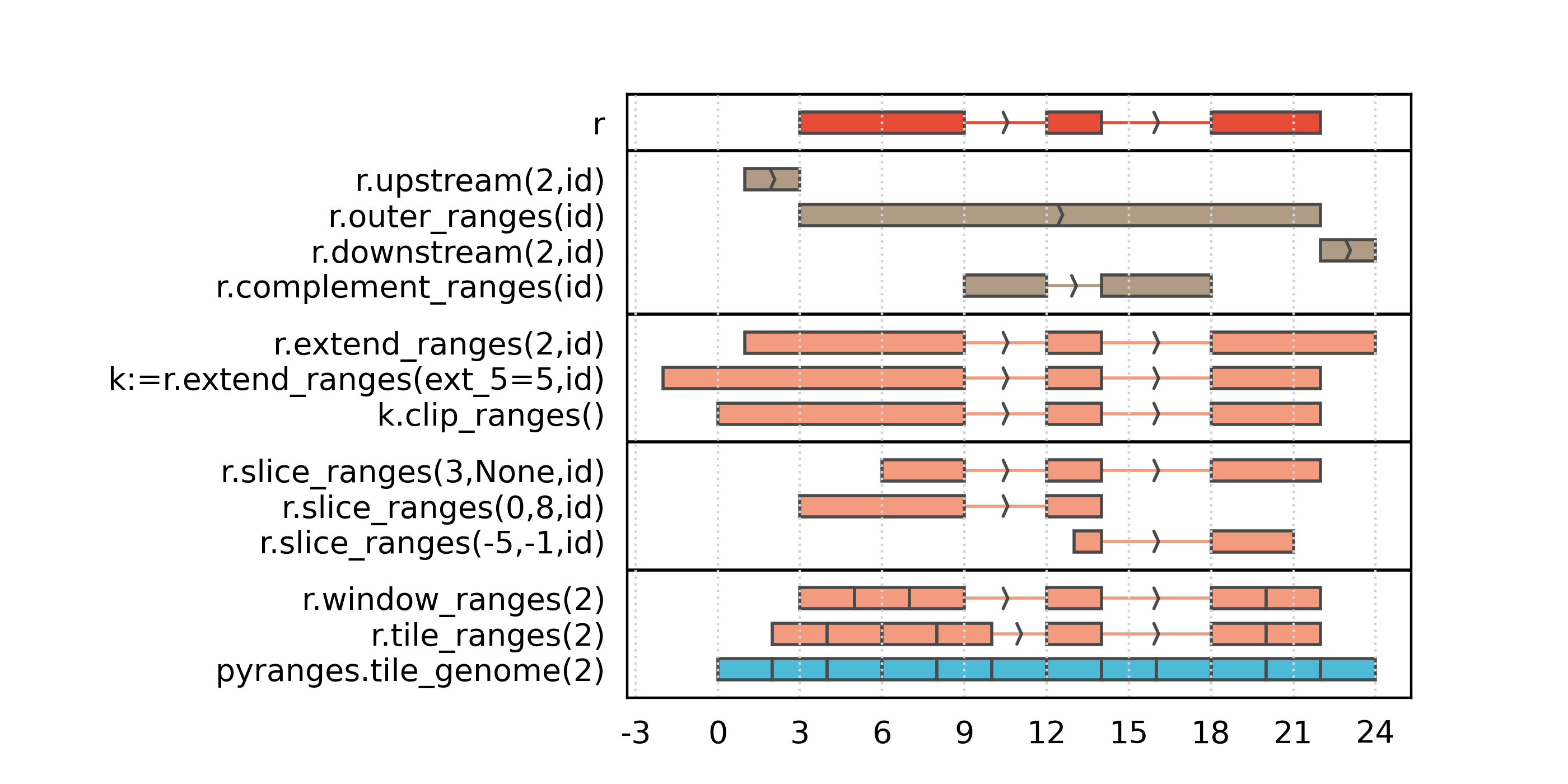
Modifying coordinates
Interval coordinates (Start, End) can be directly modified like any Series in dataframes. Let’s get some data:
>>> import pyranges1 as pr
>>> ex = pr.example_data.ensembl_gtf
>>> ex = ex[ex.Feature == "exon"].get_with_loc_columns('transcript_id')
>>> ex = ex.sort_ranges(use_strand=False).reset_index(drop=True)
>>> ex
index | Chromosome Start End Strand transcript_id
int64 | category int64 int64 category str
------- --- ------------ ------- ------- ---------- ---------------
0 | 1 11868 12227 + ENST00000456328
1 | 1 12612 12721 + ENST00000456328
2 | 1 13220 14409 + ENST00000456328
3 | 1 110952 111357 - ENST00000471248
4 | 1 112699 112804 - ENST00000471248
5 | 1 120873 120932 - ENST00000610542
6 | 1 129054 129223 - ENST00000610542
7 | 1 133373 133723 - ENST00000610542
PyRanges with 8 rows, 5 columns, and 1 index columns.
Contains 1 chromosomes and 2 strands.
We can modify a whole column at once:
>>> ex['Start'] += 5
>>> ex
index | Chromosome Start End Strand transcript_id
int64 | category int64 int64 category str
------- --- ------------ ------- ------- ---------- ---------------
0 | 1 11873 12227 + ENST00000456328
1 | 1 12617 12721 + ENST00000456328
2 | 1 13225 14409 + ENST00000456328
3 | 1 110957 111357 - ENST00000471248
4 | 1 112704 112804 - ENST00000471248
5 | 1 120878 120932 - ENST00000610542
6 | 1 129059 129223 - ENST00000610542
7 | 1 133378 133723 - ENST00000610542
PyRanges with 8 rows, 5 columns, and 1 index columns.
Contains 1 chromosomes and 2 strands.
Or we can modify a slice of the column:
>>> ex.loc[2:5, 'Start'] -= 5
>>> ex
index | Chromosome Start End Strand transcript_id
int64 | category int64 int64 category str
------- --- ------------ ------- ------- ---------- ---------------
0 | 1 11873 12227 + ENST00000456328
1 | 1 12617 12721 + ENST00000456328
2 | 1 13220 14409 + ENST00000456328
3 | 1 110952 111357 - ENST00000471248
4 | 1 112699 112804 - ENST00000471248
5 | 1 120873 120932 - ENST00000610542
6 | 1 129059 129223 - ENST00000610542
7 | 1 133378 133723 - ENST00000610542
PyRanges with 8 rows, 5 columns, and 1 index columns.
Contains 1 chromosomes and 2 strands.
Or use a boolean index:
>>> ex.loc[ex.Strand == "+", "Start"] += 5
>>> e=ex.copy()
>>> e
index | Chromosome Start End Strand transcript_id
int64 | category int64 int64 category str
------- --- ------------ ------- ------- ---------- ---------------
0 | 1 11878 12227 + ENST00000456328
1 | 1 12622 12721 + ENST00000456328
2 | 1 13225 14409 + ENST00000456328
3 | 1 110952 111357 - ENST00000471248
4 | 1 112699 112804 - ENST00000471248
5 | 1 120873 120932 - ENST00000610542
6 | 1 129059 129223 - ENST00000610542
7 | 1 133378 133723 - ENST00000610542
PyRanges with 8 rows, 5 columns, and 1 index columns.
Contains 1 chromosomes and 2 strands.
On the other hand, pyranges1 offer convenient and intuitive methods to modify coordinates, which deal
with the complexity of intervals and strands.
Next, we will showcase some of its functionalities, using the e object above as starting point.
Extending intervals
The extend_ranges method allows to extend the intervals in a PyRanges object.
The ext parameter implies an extension in both directions of all intervals:
>>> e.extend_ranges(ext=5)
index | Chromosome Start End Strand transcript_id
int64 | category int64 int64 category str
------- --- ------------ ------- ------- ---------- ---------------
0 | 1 11873 12232 + ENST00000456328
1 | 1 12617 12726 + ENST00000456328
2 | 1 13220 14414 + ENST00000456328
3 | 1 110947 111362 - ENST00000471248
4 | 1 112694 112809 - ENST00000471248
5 | 1 120868 120937 - ENST00000610542
6 | 1 129054 129228 - ENST00000610542
7 | 1 133373 133728 - ENST00000610542
PyRanges with 8 rows, 5 columns, and 1 index columns.
Contains 1 chromosomes and 2 strands.
The ext_5 and ext_3 parameters allow to specify separately the extension in the 5’ and 3’ directions,
respectively. These operations are strand-aware, meaning that a 5’ extension affects the Start position of intervals
on the positive strand, and the End position of intervals on the negative strand, and vice versa for 3’ extensions.
Let’s extend upstream by 10 bases:
>>> e.extend_ranges(ext_5=10)
index | Chromosome Start End Strand transcript_id
int64 | category int64 int64 category str
------- --- ------------ ------- ------- ---------- ---------------
0 | 1 11868 12227 + ENST00000456328
1 | 1 12612 12721 + ENST00000456328
2 | 1 13215 14409 + ENST00000456328
3 | 1 110952 111367 - ENST00000471248
4 | 1 112699 112814 - ENST00000471248
5 | 1 120873 120942 - ENST00000610542
6 | 1 129059 129233 - ENST00000610542
7 | 1 133378 133733 - ENST00000610542
PyRanges with 8 rows, 5 columns, and 1 index columns.
Contains 1 chromosomes and 2 strands.
Let’s extend by 12 bases on the 5’ end, and 6 bases on the 3’ end:
>>> e.extend_ranges(ext_5=12, ext_3=6)
index | Chromosome Start End Strand transcript_id
int64 | category int64 int64 category str
------- --- ------------ ------- ------- ---------- ---------------
0 | 1 11866 12233 + ENST00000456328
1 | 1 12610 12727 + ENST00000456328
2 | 1 13213 14415 + ENST00000456328
3 | 1 110946 111369 - ENST00000471248
4 | 1 112693 112816 - ENST00000471248
5 | 1 120867 120944 - ENST00000610542
6 | 1 129053 129235 - ENST00000610542
7 | 1 133372 133735 - ENST00000610542
PyRanges with 8 rows, 5 columns, and 1 index columns.
Contains 1 chromosomes and 2 strands.
To ignore strand (i.e. treat all intervals as if on the positive strand), use use_strand=False:
>>> e.extend_ranges(ext_5=12, ext_3=6, use_strand=False)
index | Chromosome Start End Strand transcript_id
int64 | category int64 int64 category str
------- --- ------------ ------- ------- ---------- ---------------
0 | 1 11866 12233 + ENST00000456328
1 | 1 12610 12727 + ENST00000456328
2 | 1 13213 14415 + ENST00000456328
3 | 1 110940 111363 - ENST00000471248
4 | 1 112687 112810 - ENST00000471248
5 | 1 120861 120938 - ENST00000610542
6 | 1 129047 129229 - ENST00000610542
7 | 1 133366 133729 - ENST00000610542
PyRanges with 8 rows, 5 columns, and 1 index columns.
Contains 1 chromosomes and 2 strands.
In all examples above, the extension is applied independently to all intervals in the PyRanges object.
Alternatively, you can group intervals by a column, specified with the group_by argument.
When provided, extensions are relative to the transcript, not the interval. In practice, only the first and/or last
exons of each transcript may be extended:
>>> e.extend_ranges(ext_5=10, group_by='transcript_id')
index | Chromosome Start End Strand transcript_id
int64 | category int64 int64 category str
------- --- ------------ ------- ------- ---------- ---------------
0 | 1 11868 12227 + ENST00000456328
1 | 1 12622 12721 + ENST00000456328
2 | 1 13225 14409 + ENST00000456328
3 | 1 110952 111357 - ENST00000471248
4 | 1 112699 112814 - ENST00000471248
5 | 1 120873 120932 - ENST00000610542
6 | 1 129059 129223 - ENST00000610542
7 | 1 133378 133733 - ENST00000610542
PyRanges with 8 rows, 5 columns, and 1 index columns.
Contains 1 chromosomes and 2 strands.
Slicing operations
Slicing operations are operations that cut the intervals in a PyRanges object to obtain smaller intervals. Intervals may be treated independently (default) or grouped in transcripts.
Method slice_ranges allows to
obtain slices by specifying the start and end position, in python notation.
So, to get the first 10 bases of each interval, we can do:
>>> e.slice_ranges(start=0, end=10)
index | Chromosome Start End Strand transcript_id
int64 | category int64 int64 category str
------- --- ------------ ------- ------- ---------- ---------------
0 | 1 11878 11888 + ENST00000456328
1 | 1 12622 12632 + ENST00000456328
2 | 1 13225 13235 + ENST00000456328
3 | 1 111347 111357 - ENST00000471248
4 | 1 112794 112804 - ENST00000471248
5 | 1 120922 120932 - ENST00000610542
6 | 1 129213 129223 - ENST00000610542
7 | 1 133713 133723 - ENST00000610542
PyRanges with 8 rows, 5 columns, and 1 index columns.
Contains 1 chromosomes and 2 strands.
Note above that positions refer to the 5’ end of intervals, meaning that counting
occurs from right to left for intervals on the negative strand.
You can ignore strand using use_strand=False:
>>> e.slice_ranges(start=0, end=10, use_strand=False)
index | Chromosome Start End Strand transcript_id
int64 | category int64 int64 category str
------- --- ------------ ------- ------- ---------- ---------------
0 | 1 11878 11888 + ENST00000456328
1 | 1 12622 12632 + ENST00000456328
2 | 1 13225 13235 + ENST00000456328
3 | 1 110952 110962 - ENST00000471248
4 | 1 112699 112709 - ENST00000471248
5 | 1 120873 120883 - ENST00000610542
6 | 1 129059 129069 - ENST00000610542
7 | 1 133378 133388 - ENST00000610542
PyRanges with 8 rows, 5 columns, and 1 index columns.
Contains 1 chromosomes and 2 strands.
start and end can be provided as positional arguments. end can be omitted.
When requesting a slice that is entirely out of bounds, the corresponding rows are absent in output.
The following yields intervals from position 200 to their existing 3’ end
(i.e. we remove the first 200 bases of each interval).
Note that intervals that were <200 bp have no row in output:
>>> e.slice_ranges(200)
index | Chromosome Start End Strand transcript_id
int64 | category int64 int64 category str
------- --- ------------ ------- ------- ---------- ---------------
0 | 1 12078 12227 + ENST00000456328
2 | 1 13425 14409 + ENST00000456328
3 | 1 110952 111157 - ENST00000471248
7 | 1 133378 133523 - ENST00000610542
PyRanges with 4 rows, 5 columns, and 1 index columns.
Contains 1 chromosomes and 2 strands.
Positions can be negative, in which case they are counted from the end of the interval. To get the last 10 bases of each interval, we can do:
>>> e.slice_ranges(-10)
index | Chromosome Start End Strand transcript_id
int64 | category int64 int64 category str
------- --- ------------ ------- ------- ---------- ---------------
0 | 1 12217 12227 + ENST00000456328
1 | 1 12711 12721 + ENST00000456328
2 | 1 14399 14409 + ENST00000456328
3 | 1 110952 110962 - ENST00000471248
4 | 1 112699 112709 - ENST00000471248
5 | 1 120873 120883 - ENST00000610542
6 | 1 129059 129069 - ENST00000610542
7 | 1 133378 133388 - ENST00000610542
PyRanges with 8 rows, 5 columns, and 1 index columns.
Contains 1 chromosomes and 2 strands.
This returns intervals without their first and last 3 bases:
>>> e.slice_ranges(3, -3)
index | Chromosome Start End Strand transcript_id
int64 | category int64 int64 category str
------- --- ------------ ------- ------- ---------- ---------------
0 | 1 11881 12224 + ENST00000456328
1 | 1 12625 12718 + ENST00000456328
2 | 1 13228 14406 + ENST00000456328
3 | 1 110955 111354 - ENST00000471248
4 | 1 112702 112801 - ENST00000471248
5 | 1 120876 120929 - ENST00000610542
6 | 1 129062 129220 - ENST00000610542
7 | 1 133381 133720 - ENST00000610542
PyRanges with 8 rows, 5 columns, and 1 index columns.
Contains 1 chromosomes and 2 strands.
Above, each interval is treated independently. Alternatively, you can consider transcripts,
grouping intervals (i.e. exons) by a column, specified with the group_by argument.
When provided, slice_ranges arguments are relative to the transcript, not the
interval. Note that using group_by assumes that exons belonging to the same transcript have no overlap; on the other
hand, it does not assume presorting of intervals.
By default, coordinates are relative to spliced transcripts. For example, for a transcript with two exons of 50 bp, the first position of the second exon is considered to be 50 regardless of the length of the intron in-between.
Below we request the first 1500 bases of each spliced transcript. Only exons are counted to sum up to that length, and introns are ignored:
>>> e.slice_ranges(0, 1500, group_by='transcript_id')
index | Chromosome Start End Strand transcript_id
int64 | category int64 int64 category str
------- --- ------------ ------- ------- ---------- ---------------
0 | 1 11878 12227 + ENST00000456328
1 | 1 12622 12721 + ENST00000456328
2 | 1 13225 14277 + ENST00000456328
3 | 1 110952 111357 - ENST00000471248
4 | 1 112699 112804 - ENST00000471248
5 | 1 120873 120932 - ENST00000610542
6 | 1 129059 129223 - ENST00000610542
7 | 1 133378 133723 - ENST00000610542
PyRanges with 8 rows, 5 columns, and 1 index columns.
Contains 1 chromosomes and 2 strands.
In the e object, only ENST00000456328 is larger than 1500 bases.
Compare it with the result above, noting that its third exon has been shortened:
>>> e
index | Chromosome Start End Strand transcript_id
int64 | category int64 int64 category str
------- --- ------------ ------- ------- ---------- ---------------
0 | 1 11878 12227 + ENST00000456328
1 | 1 12622 12721 + ENST00000456328
2 | 1 13225 14409 + ENST00000456328
3 | 1 110952 111357 - ENST00000471248
4 | 1 112699 112804 - ENST00000471248
5 | 1 120873 120932 - ENST00000610542
6 | 1 129059 129223 - ENST00000610542
7 | 1 133378 133723 - ENST00000610542
PyRanges with 8 rows, 5 columns, and 1 index columns.
Contains 1 chromosomes and 2 strands.
So, this will get the first and last 10 bases of each spliced transcript:
>>> first10 = e.slice_ranges(0, 10, group_by='transcript_id')
>>> last10 = e.slice_ranges(-10, group_by='transcript_id')
>>> pr.concat([first10, last10])
index | Chromosome Start End Strand transcript_id
int64 | category int64 int64 category str
------- --- ------------ ------- ------- ---------- ---------------
0 | 1 11878 11888 + ENST00000456328
4 | 1 112794 112804 - ENST00000471248
7 | 1 133713 133723 - ENST00000610542
2 | 1 14399 14409 + ENST00000456328
3 | 1 110952 110962 - ENST00000471248
5 | 1 120873 120883 - ENST00000610542
PyRanges with 6 rows, 5 columns, and 1 index columns.
Contains 1 chromosomes and 2 strands.
Subsequence operations can be combined with extensions to obtain intervals adjacent to the input ones. For example, this will obtain the 100 bases upstream of each transcript:
>>> e.extend_ranges(ext_5=100, group_by='transcript_id').slice_ranges(0, 100, group_by='transcript_id')
index | Chromosome Start End Strand transcript_id
int64 | category int64 int64 category str
------- --- ------------ ------- ------- ---------- ---------------
0 | 1 11778 11878 + ENST00000456328
4 | 1 112804 112904 - ENST00000471248
7 | 1 133723 133823 - ENST00000610542
PyRanges with 3 rows, 5 columns, and 1 index columns.
Contains 1 chromosomes and 2 strands.
This will obtain the 100 bases downstream of each transcript:
>>> e.extend_ranges(ext_3=100, group_by='transcript_id').slice_ranges(-100, group_by='transcript_id')
index | Chromosome Start End Strand transcript_id
int64 | category int64 int64 category str
------- --- ------------ ------- ------- ---------- ---------------
2 | 1 14409 14509 + ENST00000456328
3 | 1 110852 110952 - ENST00000471248
5 | 1 120773 120873 - ENST00000610542
PyRanges with 3 rows, 5 columns, and 1 index columns.
Contains 1 chromosomes and 2 strands.
However, pyranges1 provides more convenients functions to this purpose: upstream
and downstream allow to obtain regions upstream or
downstream of intervals. They allow to specify the length, as well as any optional gap between
the returned intervals and the input ones:
>>> e.downstream(100, group_by='transcript_id')
index | Chromosome Start End Strand transcript_id
int64 | category int64 int64 category str
------- --- ------------ ------- ------- ---------- ---------------
2 | 1 14409 14509 + ENST00000456328
3 | 1 110852 110952 - ENST00000471248
5 | 1 120773 120873 - ENST00000610542
PyRanges with 3 rows, 5 columns, and 1 index columns.
Contains 1 chromosomes and 2 strands.
>>> e.downstream(100, gap=10, group_by='transcript_id')
index | Chromosome Start End Strand transcript_id
int64 | category int64 int64 category str
------- --- ------------ ------- ------- ---------- ---------------
2 | 1 14419 14519 + ENST00000456328
3 | 1 110842 110942 - ENST00000471248
5 | 1 120763 120863 - ENST00000610542
PyRanges with 3 rows, 5 columns, and 1 index columns.
Contains 1 chromosomes and 2 strands.
Sometimes, you may want to slice ranges according to non-spliced coordinates. This can be done with
slice_ranges setting the count_introns argument to True.
So, the following will get the subintervals included in the first 1500 bases of each unspliced transcript:
>>> e.slice_ranges(0, 1500, group_by='transcript_id', count_introns=True)
index | Chromosome Start End Strand transcript_id
int64 | category int64 int64 category str
------- --- ------------ ------- ------- ---------- ---------------
0 | 1 11878 12227 + ENST00000456328
1 | 1 12622 12721 + ENST00000456328
2 | 1 13225 13378 + ENST00000456328
3 | 1 111304 111357 - ENST00000471248
4 | 1 112699 112804 - ENST00000471248
7 | 1 133378 133723 - ENST00000610542
PyRanges with 6 rows, 5 columns, and 1 index columns.
Contains 1 chromosomes and 2 strands.
Thus, the command above is equivalent to requesting the portions of intervals that overlap with the first 1500 bases of the boundaries of each transcript:
>>> b = e.outer_ranges('transcript_id')
>>> b
index | Chromosome Start End Strand transcript_id
int64 | category int64 int64 category str
------- --- ------------ ------- ------- ---------- ---------------
0 | 1 11878 14409 + ENST00000456328
1 | 1 110952 112804 - ENST00000471248
2 | 1 120873 133723 - ENST00000610542
PyRanges with 3 rows, 5 columns, and 1 index columns.
Contains 1 chromosomes and 2 strands.
>>> e.intersect_overlaps( b.slice_ranges(0, 1500) )
index | Chromosome Start End Strand transcript_id
int64 | category int64 int64 category str
------- --- ------------ ------- ------- ---------- ---------------
0 | 1 11878 12227 + ENST00000456328
1 | 1 12622 12721 + ENST00000456328
2 | 1 13225 13378 + ENST00000456328
3 | 1 111304 111357 - ENST00000471248
4 | 1 112699 112804 - ENST00000471248
7 | 1 133378 133723 - ENST00000610542
PyRanges with 6 rows, 5 columns, and 1 index columns.
Contains 1 chromosomes and 2 strands.
Interval complement
Another useful operation is to obtain the complement of intervals in a PyRanges object, that is,
all the bases that are not covered by any of the intervals.
This can be done with the complement_ranges method.
Let’s revise our e object:
>>> e
index | Chromosome Start End Strand transcript_id
int64 | category int64 int64 category str
------- --- ------------ ------- ------- ---------- ---------------
0 | 1 11878 12227 + ENST00000456328
1 | 1 12622 12721 + ENST00000456328
2 | 1 13225 14409 + ENST00000456328
3 | 1 110952 111357 - ENST00000471248
4 | 1 112699 112804 - ENST00000471248
5 | 1 120873 120932 - ENST00000610542
6 | 1 129059 129223 - ENST00000610542
7 | 1 133378 133723 - ENST00000610542
PyRanges with 8 rows, 5 columns, and 1 index columns.
Contains 1 chromosomes and 2 strands.
By default, the complement return includes only the internals that are not covered by any of the intervals, and split by strand; it does not include the bases before the first interval or after the last one.
>>> e.complement_ranges()
index | Chromosome Start End Strand
int64 | category int64 int64 category
------- --- ------------ ------- ------- ----------
0 | 1 12227 12622 +
1 | 1 12721 13225 +
2 | 1 111357 112699 -
3 | 1 112804 120873 -
4 | 1 120932 129059 -
5 | 1 129223 133378 -
PyRanges with 6 rows, 4 columns, and 1 index columns.
Contains 1 chromosomes and 2 strands.
Argument use_strand allows to ignore strand, returning the complement of all intervals:
>>> e.complement_ranges(use_strand=False)
index | Chromosome Start End
int64 | category int64 int64
------- --- ------------ ------- -------
0 | 1 12227 12622
1 | 1 12721 13225
2 | 1 14409 110952
3 | 1 111357 112699
4 | 1 112804 120873
5 | 1 120932 129059
6 | 1 129223 133378
PyRanges with 7 rows, 3 columns, and 1 index columns.
Contains 1 chromosomes.
A possible application of the complement operation is to obtain the intergenic regions. To do that, we use the
boundaries of each transcript group, i.e. object b obtained above:
>>> b.complement_ranges(use_strand=False)
index | Chromosome Start End
int64 | category int64 int64
------- --- ------------ ------- -------
0 | 1 14409 110952
1 | 1 112804 120873
PyRanges with 2 rows, 3 columns, and 1 index columns.
Contains 1 chromosomes.
Note that the first and last intervals are not included in the output. To do so, we set include_first_interval=True,
and provide the chromsize argument, which is a dictionary with chromosome names as keys and their sizes as values:
>>> b.complement_ranges(use_strand=False, chromsizes={'1':249250621}, include_first_interval=True)
index | Chromosome Start End
int64 | category int64 int64
------- --- ------------ ------- ---------
0 | 1 0 11878
1 | 1 14409 110952
2 | 1 112804 120873
3 | 1 133723 249250621
PyRanges with 4 rows, 3 columns, and 1 index columns.
Contains 1 chromosomes.
Another useful application of the complement operation is to obtain the coordinates of introns in a transcript.
To do this, the complement must be applied to each transcript independently, that is, grouped by a column.
This can be done with the group_by argument:
>>> introns = e.complement_ranges(group_by='transcript_id')
>>> introns
index | Chromosome Start End Strand transcript_id
int64 | category int64 int64 category str
------- --- ------------ ------- ------- ---------- ---------------
0 | 1 12227 12622 + ENST00000456328
1 | 1 12721 13225 + ENST00000456328
2 | 1 111357 112699 - ENST00000471248
3 | 1 120932 129059 - ENST00000610542
4 | 1 129223 133378 - ENST00000610542
PyRanges with 5 rows, 5 columns, and 1 index columns.
Contains 1 chromosomes and 2 strands.
Other slicing operations
Many genomic analyses involve running a sliding window over the genome or subregions of it.
Method window_ranges allows to obtain adjacent windows of a specified size and step that
span each interval in a PyRanges object.
>>> g = pr.PyRanges(dict(Chromosome=1, Start=[4, 60, 100], End=[11, 66, 107],
... Strand=['+', '+', '-'], Name=['a', 'a', 'b']))
>>> g
index | Chromosome Start End Strand Name
int64 | int64 int64 int64 str str
------- --- ------------ ------- ------- -------- ------
0 | 1 4 11 + a
1 | 1 60 66 + a
2 | 1 100 107 - b
PyRanges with 3 rows, 5 columns, and 1 index columns.
Contains 1 chromosomes and 2 strands.
For example, let’s get windows of size 3:
>>> g.window_ranges(3)
index | Chromosome Start End Strand Name
int64 | int64 int64 int64 str str
------- --- ------------ ------- ------- -------- ------
0 | 1 4 7 + a
0 | 1 7 10 + a
0 | 1 10 11 + a
1 | 1 60 63 + a
1 | 1 63 66 + a
2 | 1 104 107 - b
2 | 1 101 104 - b
2 | 1 100 101 - b
PyRanges with 8 rows, 5 columns, and 1 index columns (with 5 index duplicates).
Contains 1 chromosomes and 2 strands.
Windows are generated for each interval independently. Strand is considered: they are generated starting from the 5’
end. To ignore strand, use use_strand=False:
>>> g.window_ranges(3, use_strand=False)
index | Chromosome Start End Strand Name
int64 | int64 int64 int64 str str
------- --- ------------ ------- ------- -------- ------
0 | 1 4 7 + a
0 | 1 7 10 + a
0 | 1 10 11 + a
1 | 1 60 63 + a
1 | 1 63 66 + a
2 | 1 100 103 - b
2 | 1 103 106 - b
2 | 1 106 107 - b
PyRanges with 8 rows, 5 columns, and 1 index columns (with 5 index duplicates).
Contains 1 chromosomes and 2 strands.
To avoid duplicated indices, run pandas dataframe method reset_index on the output:
>>> g.window_ranges(3).reset_index(drop=True)
index | Chromosome Start End Strand Name
int64 | int64 int64 int64 str str
------- --- ------------ ------- ------- -------- ------
0 | 1 4 7 + a
1 | 1 7 10 + a
2 | 1 10 11 + a
3 | 1 60 63 + a
4 | 1 63 66 + a
5 | 1 104 107 - b
6 | 1 101 104 - b
7 | 1 100 101 - b
PyRanges with 8 rows, 5 columns, and 1 index columns.
Contains 1 chromosomes and 2 strands.
To may retain the old index as column, with:
>>> g.window_ranges(3).reset_index(names='g_index')
index | g_index Chromosome Start End Strand Name
int64 | int64 int64 int64 int64 str str
------- --- --------- ------------ ------- ------- -------- ------
0 | 0 1 4 7 + a
1 | 0 1 7 10 + a
2 | 0 1 10 11 + a
3 | 1 1 60 63 + a
4 | 1 1 63 66 + a
5 | 2 1 104 107 - b
6 | 2 1 101 104 - b
7 | 2 1 100 101 - b
PyRanges with 8 rows, 6 columns, and 1 index columns.
Contains 1 chromosomes and 2 strands.
To ‘window’ a whole genome (e.g. to then quantify reads in each window), pyranges1 offers
pyranges1.tile_genome(). Here, you must provide chromosome sizes, with various syntaxes accepted, and again a
window size. This function will return windows to cover all the chromosomes:
>>> cs={'chr1':323, 'chr2':125} # creating a dictionary with chromosome sizes
>>> pr.tile_genome(cs, 100)
index | Chromosome Start End
int64 | str int64 int64
------- --- ------------ ------- -------
0 | chr1 0 100
1 | chr1 100 200
2 | chr1 200 300
3 | chr1 300 323
4 | chr2 0 100
5 | chr2 100 125
PyRanges with 6 rows, 3 columns, and 1 index columns.
Contains 2 chromosomes.
Note that the last window is not full, as the chromosome size is not a multiple of the window size.
To ensure tile size consistency, use the full_last_tile parameter:
>>> pr.tile_genome(cs, 100, full_last_tile=True)
index | Chromosome Start End
int64 | str int64 int64
------- --- ------------ ------- -------
0 | chr1 0 100
1 | chr1 100 200
2 | chr1 200 300
3 | chr1 300 400
4 | chr2 0 100
5 | chr2 100 200
PyRanges with 6 rows, 3 columns, and 1 index columns.
Contains 2 chromosomes.
A related operation is tile_ranges, whose rationale is to obtain only the genome tiles (of
a defined size) that overlap the intervals in a PyRanges object:
>>> se = e.loc[[0,7],:]
>>> se
index | Chromosome Start End Strand transcript_id
int64 | category int64 int64 category str
------- --- ------------ ------- ------- ---------- ---------------
0 | 1 11878 12227 + ENST00000456328
7 | 1 133378 133723 - ENST00000610542
PyRanges with 2 rows, 5 columns, and 1 index columns.
Contains 1 chromosomes and 2 strands.
>>> se.tile_ranges(200)
index | Chromosome Start End Strand transcript_id
int64 | category int64 int64 category str
------- --- ------------ ------- ------- ---------- ---------------
0 | 1 11800 12000 + ENST00000456328
0 | 1 12000 12200 + ENST00000456328
0 | 1 12200 12400 + ENST00000456328
7 | 1 133200 133400 - ENST00000610542
7 | 1 133400 133600 - ENST00000610542
7 | 1 133600 133800 - ENST00000610542
PyRanges with 6 rows, 5 columns, and 1 index columns (with 4 index duplicates).
Contains 1 chromosomes and 2 strands.
Note that, in contrast with window_ranges, the function
tile_ranges returns intervals anchored to genome positions: their Start will always be
a multiple of the tile size, like pyranges1.tile_genome(), and regardless of the strand of the original intervals.
Argument overlap_column can be used to add a column indicating how much of the original interval
overlaps with the tile returned:
>>> se.tile_ranges(200, overlap_column='nts')
index | Chromosome Start End Strand transcript_id nts
int64 | category int64 int64 category str float64
------- --- ------------ ------- ------- ---------- --------------- ---------
0 | 1 11800 12000 + ENST00000456328 0.61
0 | 1 12000 12200 + ENST00000456328 1
0 | 1 12200 12400 + ENST00000456328 0.135
7 | 1 133200 133400 - ENST00000610542 0.11
7 | 1 133400 133600 - ENST00000610542 1
7 | 1 133600 133800 - ENST00000610542 0.615
PyRanges with 6 rows, 6 columns, and 1 index columns (with 4 index duplicates).
Contains 1 chromosomes and 2 strands.